Lettre à communauté AS
Recrutement TANGELO terminé et annonce de l’essai clinique portant sur l’Alogabat
07 décembre 2022
Chère communauté mondiale du syndrome d’Angelman,
Dans le cadre de notre engagement continu à partager des mises à jour importantes et opportunes sur notre programme dans le syndrome d’Angelman (SA), nous sommes heureux d’annoncer que tous les participants de la première partie ou la partie Multiple Ascending Dose (MAD) de l’étude TANGELO ont réussi et sont désormais inscrits dans la partie d’extension à long terme (LTE) de TANGELO. Cette partie évaluent maintenant différents niveaux de dose avec des intervalles plus longs entre les administrations pour une période prolongée pour examiner l’innocuité et la tolérabilité à long terme de Rugonersen.
De plus, en témoignage de notre engagement envers SA, nous sommes heureux d’annoncer que nous élargissons notre portefeuille pour soutenir davantage le développement d’options de traitement dans le SA.
S’appuyant sur l’important travail réalisé avec la communauté SA, comme le modèle conceptuel de la maladie, l’étude observationnelle FREESIAS et de nombreuses études précliniques, collaborations avec des partenaires académiques, ainsi que nos recherches en cours avec Rugonersen, nous élargissons notre portefeuille de développement de médicaments dans le SA à Alogabat, un modulateur du GABA qui est pris sous forme de comprimé oral une fois par jour. Suite aux informations partagées lors du congrès FAST 2022 Industry Update, nous prévoyons une nouvelle étude clinique qui explorera le potentiel de l’Alogabat comme traitement pour les individus SA avec un génotype de délétion.
Cela reflète notre objectif de trouver des traitements potentiels pour les troubles neurogénétiques rares comme la SA. Nous croyons qu’il est important de développer un certain nombre d’approches différentes pour lutter contre les maladies complexes comme SA. Le ciblage du SA de plusieurs manières, et potentiellement complémentaires, peut augmenter la chances de développer un traitement efficace et, en fin de compte, d’améliorer les soins aux patients.
Pourquoi investiguons nous une nouvelle piste avec l’Alogabat ?
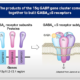
Le SA est une maladie complexe et hétérogène causée par une perturbation du gène UBE3A, qui est vital pour la façon dont le cerveau contrôle la parole, le mouvement et l’apprentissage.
La recherche montre que les personnes atteintes de délétion ont une présentation plus sévère que les autres SA génotypes. Cela suggère que d’autres gènes au-delà d’UBE3A, lorsqu’ils sont supprimés, peuvent contribuer à des symptômes plus graves.
Certains de ces gènes, situés à proximité du gène UBE3A sur le même chromosome 15, sont appelés GABRB3, GABRA5 et GABRG3 et sont délétés maternellement chez les individus avec génotype de type délétion. Ces trois gènes contiennent l’information pour coder pour trois protéines qui s’assemblent pour former les récepteurs GABAA α5.
Un récepteur comme le récepteur GABAA α5 est une protéine qui réside sur les cellules du cerveau et qui fonctionne comme un trou de serrure où la clé est un neurotransmetteur ou une substance utilisée par le cerveau pour communiquer au sein de ses cellules. La clé qui est reconnue par le trou de serrure GABAA α5 est un neurotransmetteur appelé GABA. GABA et les récepteurs GABAA α5 jouent un rôle important dans le développement du cerveau, le sommeil, le comportement et le contrôle des crises d’épilepsie, et parce que la quantité de GABAA α5 est réduit chez les individus SA avec délétion, ils peuvent jouer un rôle dans les cas les plus graves des symptômes observés chez ces personnes
Qu’est-ce qu’Alogabat ?
L’Alogabat est une molécule connue comme modulateur allostérique positif c’est-à-dire qu’il améliore l’activité des récepteurs GABAA α5 en présence du neurotransmetteur GABA. Alogabat peut restaurer le fonctionnement du GABA chez les individus SA avec un génotype de type délétion et peut améliorer certains des symptômes et potentiellement aider au développement du cerveau. Cela a le potentiel de susciter un bénéfice chez les patients délétés indépendamment et en plus du bénéfice attendu des traitements visant uniquement à restaurer la fonction UBE3A.
A quoi ressemblera l’étude Alogabat ?
L’étude alogabat est une étude de Phase IIa. La première partie de l’étude portera sur les la pharmacocinétique ou comment se comporte le médicament au sein du le corps, et l’innocuité et la tolérabilité de l’Alogabat.
La deuxième partie de l’étude est une preuve de mécanisme qui testera différentes doses d’Alogabat et son effet sur l’électroencéphalogramme (EEG).
Les détails de l’étude (numéro d’essai : NCT05630066) sont les suivants :
- L’étude est une étude ouverte, ce qui signifie que tous les participants recevront Alogabat
- Les participants recevront de l’Alogabat sous forme de comprimé oral pris une fois par jour pendant 12 semaines
- Il est prévu que l’étude soit ouverte aux enfants et adolescents (âgés de 5 à 17 ans) atteints du SA génotype de type délétion
- L’étude devrait être menée dans 6 pays au maximum
- Notre objectif est de commencer à recruter au premier semestre 2023
Nous partagerons plus de détails sur l’étude dès que possible. En attendant, nous avons développé un court document de Questions-Réponses à la fin de cette lettre qui couvre certains des points clés.
Qu’est-ce que cela signifie pour la communauté SA ?
L’annonce d’aujourd’hui est une étape importante. Il ouvre une nouvelle voie de recherche et nous rapproche un peu plus de notre objectif ultime de fournir de futurs traitements efficaces pour le SA. Comme nous le savons tous, développer un nouveau médicament est un processus long et complexe. Le progrès dans la recherche n’est possible qu’avec votre coopération continue et nous vous en sommes extrêmement reconnaissants du soutien de la communauté SA. Ensemble, nous pouvons travailler à faire avancer la science, les connaissances sur le SA et améliorer la vie des personnes atteintes du SA.
Merci encore pour votre soutien continu et si vous avez des questions sur cette annonce, n’hésitez pas à nous contacter.
Sincèrement,
Brenda Vincenzi, M.D.
Directeur médical principal
DP Neurosciences
Shady Sédhom
Directeur du partenariat mondial avec les patients
Questions-Réponses
A qui sera ouvert l’essai clinique Alogabat ?
L’essai sera ouvert aux enfants et adolescents avec un génotype de type délétion SA âgés de 5 à 17 ans.
Quand commencera-t-il ?
Nous menons actuellement des discussions avec les autorités sanitaires pour finaliser les détails de l’étude et nous visons un démarrage au premier semestre 2023.
Où se déroulera l’essai ?
L’étude devrait être menée dans 6 pays au maximum.
Où puis-je trouver plus d‘informations sur l’essai ?
Plus d’informations peuvent être trouvées dans clinicaltrial.gov et forpatients.com.
Comment puis-je m‘assurer que mon enfant est inscrit à l’essai ?
Si vous pensez que votre enfant peut répondre aux critères d’inclusion de l’essai, une fois que les détails de l’étude ont été annoncés et que l’étude commence le recrutement, vous devez parler au médecin de votre enfant ou spécialiste.
Comment les centres investigateurs seront-ils sélectionnés ?
Plusieurs facteurs seront pris en compte lors du choix de nos sites d’étude pour l’Alogabat. Ceux-ci inclus lois et réglementations internationales, nationales et locales applicables et le niveau d’expérience du site potentiel sur des études SA. Nous examinerons également l’infrastructure et la capacité du site potentiel qui doit mener l’étude parallèlement à ses activités habituelles sur le site, à quelle vitesse et efficacement, ils peuvent lancer l’étude, la population SA locale et géographique et les facteurs d’accès.
Roche envisage-t-il d‘autres essais cliniques sur l’Alogabat ?
Il s’agit du premier essai que nous allons mener avec Alogabat pour les patients atteints du SA. En fonction des résultats, nous envisagerons la possibilité de mettre en place d’autres essais.
Poursuivez-vous encore des essais avec Rugonersen dans SA ?
Oui. Nous allons poursuivre notre programme de développement clinique avec Rugonersen. Les informations peuvent être trouvées via le site Web clinicaltrial.gov et notre site Web ForPatients.
Envisagez-vous de planifier d’autres essais cliniques à Rugonersen uniquement en 2026 ?
À la lumière du profil d’innocuité et de tolérabilité acceptable du Rugonersen, ainsi que des signaux prometteurs préliminaires, nous analyserons l’ensemble de données, y compris les données entrantes de la phase d’extension à long terme dans les mois à venir en préparation des prochaines étapes avec l’objectif de poursuivre le développement clinique du Rugonersen avant 2026.
| Disclaimer : FAST France met à disposition de la communauté francophone cette traduction issue du communiqué de Roche que vous pouvez retrouver ici. En cas de divergence imputable à une erreur de traduction ou de compréhension, uniquement les sources originelles en anglais prévalent. En aucun cas, FAST France ne pourra être tenu pour responsable de ces erreurs. |