Les tests génétiques dans le cas du syndrome d’Angelman
La génétique permet dans environ 90% des cas d’établir avec certitude le diagnostic sur le syndrome d’Angelman.
Le schéma ci-dessous reprend les différentes analyses génétiques réalisées dans le syndrome.
Si un certain nombre de termes ne vous sont pas compréhensibles (analyse chromosomique, test de FISH, test de méthylation, test PCR, séquençage de l’UBE3A) alors nous vous invitons à lire les sections associées.

Traduction :
- AS : Angelman Syndrome
- UPD : Disomie uniparentale
- ID : Défaut d’empreinte
- Le syndrome Prader-Willi est une autre maladie génétique localisée sur la région 15q11-q13 du père et non de la mère comme dans le cas du syndrome d’Angelman.
Ceci est une explication des tests génétiques disponibles pour les causes connues du syndrome d’Angelman. Cet article est écrit pour les parents et les profanes qui n’ont pas une connaissance pratique de la génétique ou de la biologie moléculaire mais qui aimeraient comprendre les détails plus techniques de ces tests.
Il existe de nombreux tests pour diagnostiquer le syndrome d’Angelman. Si vous essayez de comprendre les résultats des tests de votre enfant ou de quelqu’un d’autre, il est important de savoir exactement quel(s) test(s) votre enfant a déjà passé.
Il est courant que les parents sachent que leur enfant a été testé pour le SA et se souviennent que le généticien leur a dit que le test était positif ou négatif, mais il existe plusieurs anomalies génétiques qui peuvent provoquer le SA et chaque test ne détecte qu’un certain nombre de cas.
Si un enfant a déjà été testé et que les résultats étaient négatifs, il est important de savoir quels tests ont été effectués pour savoir si le SA a vraiment été exclue.
De plus, environ une personne sur dix qui présente tous les signes cliniques du syndrome d’Angelman a des résultats normaux ou «négatifs» à tous ces tests. Ces personnes peuvent recevoir un diagnostic «clinique» du SA. Cela signifie que l’individu répond aux critères de diagnostic du SA, mais nous ne savons pas quel type d’anomalie génétique est à l’origine de ses symptômes. Rappelez-vous que toutes les personnes atteintes du SA avaient des diagnostics «cliniques» jusqu’au début des années 1990, date à laquelle les premiers tests ont été développés pour tester le chromosome 15 pour les délétions. Des recherches plus poussées sur la génétique du syndrome d’Angelman donneront probablement plus de causes du SA et, par conséquent, davantage de tests pour confirmer le diagnostic clinique.
Analyse chromosomique standard ou analyse cytogénétique
Toute femme enceinte ayant subi un prélèvement de villosités choriales (CVS encore appelé biopsie de trophoblaste) ou une amniocentèse pendant une grossesse a eu une version de ce test.
Ces tests standards recherchent des changements évidents dans la structure des chromosomes et peuvent détecter des syndromes lorsque des délétions, réarrangements ou duplications extrêmement importants se produisent. Par exemple, dans le syndrome de Down (ou trisomie 21), un fragment supplémentaire du chromosome 21 est présent et peut être vu sur le caryotype généré avec ce test.
Ce n’est pas un test détaillé et ne révèle que rarement de petites erreurs chromosomiques comme les délétions AS courantes. Ce test chromosomique standard est utile pour écarter les syndromes qui pourraient sembler similaires au SA chez le jeune enfant. Les futurs parents doivent noter que les échantillons prélevés pour les tests de caryotype standard peuvent être utilisés pour l’analyse FISH afin de détecter des syndromes spécifiques. Actuellement, le dépistage du syndrome d’Angelman n’est pas systématiquement inclus dans les tests prénataux car le syndrome est si rare.
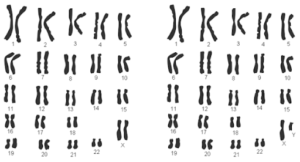
Ce schéma représente deux caryotypes, ou carte des chromosomes humains. Vous pouvez voir chaque ensemble de chromosomes correspondant du plus grand (chromosome 1) au plus petit (chromosome 22) et les chromosomes xx caractérisant une femme et xy caractérisant un homme.
 Si nous pensons à nos chromosomes comme un ensemble d’encyclopédies, alors l’analyse chromosomique standard revient à aligner l’ensemble des volumes de l’encyclopédie dans l’ordre numérique. Nous pouvons considérer chaque chromosome comme un «volume» dans une encyclopédie avec deux copies de chaque volume/chromosome. Il convient de s’assurer qu’il y a deux exemplaires de chaque chromosome sans «volumes» supplémentaires ou manquants.
Si nous pensons à nos chromosomes comme un ensemble d’encyclopédies, alors l’analyse chromosomique standard revient à aligner l’ensemble des volumes de l’encyclopédie dans l’ordre numérique. Nous pouvons considérer chaque chromosome comme un «volume» dans une encyclopédie avec deux copies de chaque volume/chromosome. Il convient de s’assurer qu’il y a deux exemplaires de chaque chromosome sans «volumes» supplémentaires ou manquants.
Le test de méthylation de l’ADN
Ce test peut également être appelé test PCR spécifique de « méthylation d’hybridation Southern » ou test PCR spécifique de méthylation. Le mot clé est « méthylation » et c’est le test de diagnostic le plus sensible du syndrome d’Angelman.
Ce test identifiera positivement environ 80% des personnes atteintes du syndrome d’Angelman. La méthylation fait référence à une «étiquette» chimique ajoutée à l’ADN et peut être utilisée pour déterminer si l’ADN a été fourni par la mère ou le père. L’endroit où cette balise est ajoutée se produit selon un modèle distinct s’il provient de la mère ou s’il provient du père.
Par conséquent, le motif de cette étiquette peut être examiné pour déterminer quel parent a contribué à l’ADN. Le test de méthylation de l’ADN déterminera si un individu a une copie de chaque modèle (un de chaque parent) ou une anomalie du syndrome d’Angelman (où seul le modèle du père est présent.)
En utilisant notre analogie d’encyclopédie, l’ADN est comme les pages à l’intérieur des volumes d’une encyclopédie. Le test de méthylation revient à ouvrir les deux «volumes» du chromosome 15 et à consulter la page du «chapitre» spécifique fourni par chaque parent pour voir quel modèle parent est visible sur la page.
Voir l’illustration ci-dessous pour découvrir une façon dont les scientifiques déterminent quels «chapitres» sont présents dans les chromosomes d’un individu. Si ce test révèle que le modèle maternel distinct est absent du chapitre spécial, l’individu testé a le syndrome d’Angelman et des tests supplémentaires sont nécessaires pour déterminer si cela est causé par une délétion, une disomie uniparentale (UPD) ou un défaut d’empreinte (ICD).
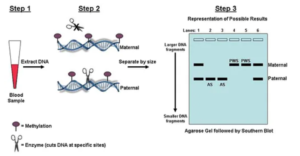
C’est une façon pour les scientifiques de déterminer quels modèles de méthylation sont présents sur le chromosome 15. Il y a d’autres façons de faire ce test au niveau technique, mais cet exemple est pour vous montrer à quoi pourrait ressembler un résultat.
Étape 1 – Un échantillon de sang est prélevé sur l’individu à tester. L’ADN des cellules sanguines est isolé.
Étape 2 – L’ADN de la première étape contient tout l’ADN de l’individu, y compris les deux copies du chromosome 15. Les groupes méthyle sont des marqueurs chimiques ajoutés à l’ADN. Ces «étiquettes» de groupe méthyle existent sur chaque «volume» du chromosome 15 où l’on s’attendrait à voir le modèle distinct associé uniquement à la mère ou uniquement au père. Les endroits de l’ADN où les groupes méthyle sont ajoutés dépendent du parent d’où provient le chromosome. Notez dans le diagramme que la copie maternelle a un nombre et un placement différents de groupes méthyle (représentés par des ovales violets) par rapport à la copie paternelle. L’échantillon d’ADN est coupé en petits morceaux avec des enzymes. Une enzyme est une protéine qui agit comme un ciseau et ne coupe l’ADN que dans des régions spécifiques. Dans la copie maternelle, un site spécifique est bloqué par le groupe méthyle et n’est pas accessible par les ciseaux. La copie paternelle peut être coupée car cette zone n’est pas cachée à l’enzyme.
Étape 3 – Les morceaux d’ADN générés à l’étape 2 sont séparés par taille sur un gel d’agarose. Un gel d’agarose ressemble beaucoup à un morceau de « Jello » qui a durci. L’ADN peut être ajouté au sommet du gel et forcé de se déplacer à travers le gel en appliquant de l’électricité sur le gel. L’ADN a une charge négative et se déplacera vers une source électrique positive. Puisque les morceaux d’ADN doivent trouver leur chemin autour des particules de Jello, les morceaux plus petits se déplacent plus rapidement que les plus grands, ainsi les morceaux plus petits se rapprochent du fond du gel. Notez à l’étape 2 que la copie paternelle sera coupée tandis que la copie maternelle ne sera pas coupée, ainsi la copie paternelle produira un plus petit morceau d’ADN. Les chercheurs peuvent ensuite utiliser une procédure connue sous le nom de «Southern blot» (ou transfert d’ADN) pour visualiser uniquement les morceaux d’ADN de la zone Angelman / Prader-Willi du chromosome 15.
Les résultats représentatifs sont visibles sur l’illustration ci-dessus. Dans les pistes 1 et 6, la copie maternelle (bande noire plus grande) et la copie paternelle (bande noire plus petite) sont présentes sur le gel, ainsi ces individus ont une méthylation correcte sur leurs copies du chromosome 15. Dans les lignes 2 et 3, les individus n’ont que le schéma de méthylation paternel présent, donc ces individus n’ont pas le schéma de méthylation maternel et ont le syndrome d’Angelman (SA). À titre de comparaison, les individus des couloirs 4 et 5 n’ont que le modèle de méthylation maternel et n’ont pas le modèle paternel. Ces personnes sont atteintes du syndrome de Prader-Willi (PWS).
Le test FISH
Le FISH signifie «hybridation fluorescente in situ». Ce test détermine si une partie du chromosome 15 est physiquement absente de l’individu.
Ce test revient à compter les pages des deux «volumes» du Chromosome 15 et à détecter les pages manquantes dans le volume. S’il y a trop peu de pages dans l’un des volumes, c’est comme si une section avait été arrachée, et nous savons que certaines des «pages» ont été supprimées. Ce test ne peut pas nous dire le chromosome du parent sur lequel il manque des pages, donc un test FISH doit être effectué avec le test de méthylation de l’ADN pour confirmer que l’individu a le syndrome d’Angelman et non le syndrome de Prader-Willi, un trouble causé par l’absence d’un «chapitre» du chromosome du père plutôt que celui de la mère.
Si un individu a un test de méthylation positif pour le SA et un test FISH positif pour la perte de chromosome 15, alors nous savons que la personne a le syndrome d’Angelman causé par une délétion. C’est la cause la plus fréquente du SA et environ 70% des personnes atteintes du SA ont cette délétion. Ces délétions peuvent survenir au hasard des mois avant même la naissance de la mère. Les ovules d’une femme se forment alors qu’elle est elle-même fœtus à environ cinq mois de gestation. Les chromosomes dans ses œufs en développement se dupliquent et se séparent rapidement à mesure que les œufs se multiplient, et des anomalies dans la structure des chromosomes peuvent facilement se produire à ce stade. Ces anomalies de cellule sont courantes et généralement inoffensives. La plupart des œufs contenant des anomalies, s’ils sont fécondés, ne se développent pas et n’entraînent pas de grossesse. Certaines délétions sont inoffensives et peuvent donner un bébé parfaitement normal. Les délétions sont un problème dans le petit nombre de cas où le chromosome paternel qui fécondera plus tard l’ovule ne peut pas compenser les informations manquantes. C’est ce qui se produit dans le syndrome d’Angelman. Les délétions chromosomiques sont relativement courantes et il n’y a aucune preuve que quelque chose de spécifique les a provoquées ou aurait pu les empêcher; c’est simplement aléatoire.
Si le test FISH est positif mais que le test de méthylation est négatif pour le syndrome d’Angelman, alors l’individu est atteint du syndrome de Prader-Willi, un trouble distinctement causé par le même mécanisme d ‘«empreinte». Chez ces individus, la délétion s’est produite sur le chromosome paternel, c’est comme si un «chapitre» manquait dans le volume du père pour lequel le chromosome de la mère ne peut pas compenser. Dans le test de méthylation, ces individus ne montreront que le schéma de méthylation de la mère et n’ont pas le schéma du père.
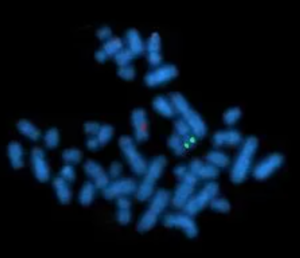
Ceci est une image réelle des chromosomes qui ont été testés en utilisant le test FISH. Pour effectuer ce test pour le SA, une sonde pour le gène UBE3A est générée. La sonde est une séquence qui trouvera sa correspondance exacte sur le chromosome 15 et se liera uniquement au gène UBE3A. La sonde est également «étiquetée» avec une molécule fluorescente afin qu’elle puisse être vue sur les chromosomes au microscope. L’image ci-dessus montre un résultat de test FISH positif pour le syndrome d’Angelman. Dans ce test, deux sondes ont été marquées avec des molécules fluorescentes; une sonde est marquée en vert et une en rouge. Un gène (utilisé comme contrôle) commun sur les deux chromosomes a été marqué avec la molécule fluorescente verte; les deux points verts confirment la présence de deux copies de ce gène dans l’échantillon, ce qui montre que les deux chromosomes possèdent le gène que l’étiquette verte testait. La sonde faite pour trouver UBE3A a été marquée avec une molécule fluorescente rouge mais il n’y a qu’un seul point rouge sur cette image. Il s’agit d’un résultat anormal et signifie qu’une copie d’UBE3A est absente de l’ADN de cette personne. Ce résultat de laboratoire ne peut pas nous dire si le chromosome avec l’étiquette rouge provient de la copie maternelle ou paternelle du chromosome 15, mais c’est un résultat positif indiquant que cette personne a une délétion sur l’un de ses deux chromosomes 15. Le test de méthylation de l’ADN est nécessaire pour confirmer si cette personne est atteinte du syndrome d’Angelman ou du syndrome de Prader-Willi.
Test PCR pour détecter la disomie uniparentale (UPD) et les défauts du centre d’impression (ICD)
Si un individu a un test de méthylation positif pour le syndrome d’Angelman, mais un test FISH négatif, alors il a soit UPD ou ICD. Un test de PCR (réaction en chaîne par polymérase) est ensuite utilisé pour déterminer si l’individu a deux copies du chromosome 15 du père (UPD) ou si l’individu a un chromosome de chaque parent, mais avec une méthylation incorrecte (ICD). Ce test nécessite un échantillon de sang des parents ainsi que de l’individu afin que l’information génétique propre à chaque parent puisse être recherchée dans les chromosomes de l’enfant.
C’est comme prendre les deux «volumes» de Chromosome 15 et rechercher des sections fournies par chaque parent spécifique. Il recherche des informations qui seraient présentes dans les deux volumes, ainsi que des informations que seul le «volume» du père et seul le «volume» de la mère contiendraient. Si le test révèle que les deux «volumes» ont été hérités du père et qu’aucun n’a été hérité de la mère, alors le test est positif pour le syndrome d’Angelman causé par UPD. L’UPD est une occurrence aléatoire au moment de la conception où l’œuf perd la copie du chromosome 15 de la mère et la copie du père se duplique pour compenser cette absence. Rien n’est connu pour le provoquer ou pour l’empêcher, c’est simplement aléatoire.
Si le test PCR montre que les deux parents ont contribué au «volume» 15 exactement comme la nature le voulait, ce résultat indique que l’individu doit avoir un ICD (défaut d’empreinte). Chez les personnes ayant un ICD, même si l’individu a hérité d’une copie du chromosome 15 de la mère et que l’UBE3A est présent, le schéma de méthylation n’est pas correctement établi. Un ICD, c’est comme avoir une erreur dans la «table des matières» du 15ème chromosome de la mère. Le volume est complet mais les cellules ne peuvent pas trouver les informations dont le cerveau a besoin à cause d’une «faute de frappe» dans la table des matières. Le défaut d’empreinte peut être examiné plus en détail pour comprendre la nature de cette «faute de frappe». La faute de frappe peut être une minuscule suppression, comme si une section de la table des matières avait été effacée, ou une mutation comme si les pages étaient «renumérotées» ou brouillées afin que les cellules du cerveau ne puissent pas trouver les pages dont elles ont besoin. Certains cas d’ICD sont héréditaires, ce qui signifie que la mère avait cette erreur dans sa propre «table des matières», et des tests supplémentaires de la mère sont réalisés pour voir si elle porte cette erreur. Si cette «faute de frappe» était aléatoire et n’est pas présente dans le «volume» de la mère, alors ses chances d’avoir un autre enfant atteint de SA sont très faibles. Mais si la mère elle-même a cette «faute de frappe», alors ses chances d’avoir un autre enfant avec la même faute de frappe sont d’au moins 50%. Pour comprendre comment une mère peut porter cette «faute de frappe» et ne pas avoir elle-même de symptômes du SA, rappelez-vous que si la mère avait hérité de cette faute de frappe de son père, cela aurait été silencieux en elle et ne lui aurait pas causé de problèmes. Pour plus d’exemples, regardez les scénarios génétiques sous séquençage UBE3A.
Séquençage UBE3A
Environ 20% des personnes qui présentent tous les symptômes du syndrome d’Angelman obtiendront des résultats négatifs à tous les tests répertoriés jusqu’à présent. Le test suivant consiste à séquencer directement leur gène UBE3A.
Le séquençage, c’est comme ouvrir le chromosome 15 hérité de la mère et regarder le «chapitre» UBE3A et vérifier soigneusement l’orthographe de chaque mot. C’est plus compliqué qu’il n’y paraît. Chacun de nous a de légères différences dans la façon dont les «mots» de notre ADN sont orthographiés, mais ces différences ne changent pas la signification de l’information et n’affectent pas la façon dont nous apprenons ou nous développons. Par exemple, le mot «color» peut également être orthographié «colour» et les deux sont corrects. Les changements qui se produisent dans l’ADN qui ne semblent pas affecter la fonction des gènes sont appelés polymorphismes; ce sont des différences inoffensives qui se produisent naturellement entre les personnes. Si un individu a un changement dans la séquence de son gène UBE3A, il est important de déterminer si ce changement est un polymorphisme inoffensif ou une mutation réelle qui affecte la capacité du gène à fonctionner correctement et qui a causé le syndrome d’Angelman chez la personne testée.
Pour comprendre comment fonctionne ce test, vous devez comprendre comment votre ADN est utilisé pour produire des protéines telles que UBE3A. La séquence d’ADN est composée de nucléotides de 4 types : A (adénine), C (cytosine), G (guanine) et T (thymine). Ces quatre «lettres» sont liées ensemble dans différentes combinaisons pour produire des chromosomes. L’ADN est un «double brin» et ressemble à une échelle tordue. Les nucléotides forment les «échelons» de l’échelle en se liant les uns aux autres. A se lie à T. C se lie à G.
La séquence de A, C, G et T sur vos chromosomes peut être lue par vos cellules tout comme vous pouvez lire un livre. Lorsqu’une séquence fournit les instructions nécessaires pour produire une protéine, nous appelons cette séquence un gène. Ainsi, le gène UBE3A est une séquence de A, C, G et T sur le chromosome 15 qui indique à la cellule comment fabriquer la protéine UBE3A. Pour fabriquer la protéine, votre cellule «lit» l’ADN et produit une copie des informations importantes dans un brin d’ARN. Dans l’ARN, la séquence est la même que celle de l’ADN, mais le nucléotide T est remplacé par U (uracile). Vous pouvez considérer l’ARN comme une page importante de l’encyclopédie que vous photocopiez pour l’emporter ailleurs. La cellule «lit» alors l’ARN en regardant les nucléotides par groupes de trois. Ces mots de trois lettres sont appelés code génétique et indiquent à la cellule quels acides aminés doivent être placés les uns à côté des autres afin de créer une protéine fonctionnelle.
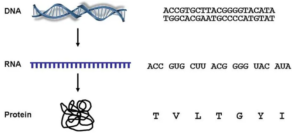 Cette figure illustre comment l’ADN fournit les informations de séquences nécessaires pour fabriquer des protéines. L’ADN est double brin et composé des quatre nucléotides A, C, G et T. Notez que dans les deux brins, A s’apparie toujours avec T et C s’apparie toujours avec G. La cellule peut copier la séquence d’ADN nécessaire pour fabriquer une protéine en une molécule simple brin appelée ARN. Notez que l’ARN dans cet exemple est exactement le même que le brin supérieur d’ADN, sauf que le nucléotide T est remplacé par U. L’ARN est lu par la cellule comme des «mots» de trois lettres. Chaque «mot» indique à la cellule quel acide aminé doit être ajouté dans quel ordre pour former une protéine fonctionnelle. Dans l’exemple ci-dessus, le «mot» CUU indique à la cellule d’ajouter l’acide aminé Leucine (L) à côté de l’acide aminé Valine (V) qui a été codé par le «mot» GUG.
Cette figure illustre comment l’ADN fournit les informations de séquences nécessaires pour fabriquer des protéines. L’ADN est double brin et composé des quatre nucléotides A, C, G et T. Notez que dans les deux brins, A s’apparie toujours avec T et C s’apparie toujours avec G. La cellule peut copier la séquence d’ADN nécessaire pour fabriquer une protéine en une molécule simple brin appelée ARN. Notez que l’ARN dans cet exemple est exactement le même que le brin supérieur d’ADN, sauf que le nucléotide T est remplacé par U. L’ARN est lu par la cellule comme des «mots» de trois lettres. Chaque «mot» indique à la cellule quel acide aminé doit être ajouté dans quel ordre pour former une protéine fonctionnelle. Dans l’exemple ci-dessus, le «mot» CUU indique à la cellule d’ajouter l’acide aminé Leucine (L) à côté de l’acide aminé Valine (V) qui a été codé par le «mot» GUG.
Il y a de nombreux endroits dans tous nos gènes qui peuvent avoir des différences d’ADN aléatoires ou des fautes d’orthographe qui n’affectent en rien la façon dont nous apprenons ou nous développons. Certaines de ces différences sont des variations naturelles (comme «color» et «colour») tandis que d’autres sont des fautes d’orthographe trop mineures pour affecter le sens de la phrase. Si nous regardions attentivement l’ADN ou le «texte» dans chacun de nos chromosomes, nous trouverions toutes sortes de «mots» mal orthographiés. Toutes les différences de séquence d’ADN ne sont pas égales, tout comme une seule faute d’orthographe dans une phrase peut être insignifiante ou peut complètement changer le sens de la phrase. Il est difficile de simplement regarder le gène UBE3A et de savoir à quel point une faute d’orthographe est significative. Pour comprendre les types de différences qui peuvent survenir et ce qu’elles peuvent signifier, veuillez consulter le diagramme suivant où nous comparons des séquences et des changements avec des phrases d’un livre.

Sur la gauche se trouve notre exemple de séquence et la protéine qu’elle coderait. Sur la droite se trouvent notre exemple d’encyclopédie et nos phrases.
- Dans le premier exemple de changement de séquence (n ° 1), ce changement est un polymorphisme et n’affecte pas la protéine produite. Vous pouvez voir que les deux «mots» ACC et ACG signifient Thréonine pour la cellule. Donc, même s’il y a un changement, cela n’affecte pas la protéine. Dans notre analogie avec l’encyclopédie, la phrase est légèrement modifiée, mais le sens est exactement le même.
- Dans le deuxième exemple de changement de séquence (n ° 2), ce changement a changé la signification de la séquence et a changé la protéine. Dans ce cas, le mot signifiant Leucine (L) a changé pour signifier le mot Proline (P). Cela peut modifier considérablement la protéine et la rendre non fonctionnelle ou moins fonctionnelle. Dans notre analogie de phrase, vous pouvez voir qu’un mot a changé et que la phrase n’a plus de sens.
- Dans le troisième exemple (n ° 3), un changement dans la séquence indique à la cellule d’arrêter de produire la protéine prématurément. Normalement, la séquence «stop», dans ce cas UAA, ne se trouve qu’à la toute fin de la protéine. Ce changement de séquence a ajouté cette instruction d’arrêt trop tôt et la protéine entière n’est pas produite. Dans notre analogie, c’est comme mettre le point final dans la phrase qui arrête la phrase trop tôt et cela n’a plus de sens.
- Dans le quatrième exemple (n ° 4), un changement de séquence s’est produit lorsqu’un nucléotide supplémentaire a été ajouté dans la séquence. Puisque nous savons que les cellules lisent la séquence en mots de trois lettres, cela décale les mots et en crée de nouveaux à partir du code d’origine. La même chose peut se produire si des délétions d’un ou plusieurs nucléotides sont trouvées. Tous les ajouts ou suppressions qui modifient l’espacement des mots à trois lettres feront probablement une protéine non fonctionnelle. Dans notre analogie des phrases, l’espacement a changé et maintenant les mots n’ont plus de sens.
Dans l’exemple n ° 1, ce ne serait pas un changement qui cause le syndrome d’Angelman. Dans l’exemple n ° 3, ce changement provoquerait très probablement le syndrome d’Angelman car les protéines qui ne sont pas complètement fabriquées sont généralement non fonctionnelles. De même, l’exemple n ° 4 serait la confirmation d’un diagnostic de syndrome d’Angelman. Les changements de l’exemple n ° 2 sont plus difficiles à déterminer s’ils sont nuisibles ou non. Dans l’exemple fourni, ce changement provoquerait probablement le syndrome d’Angelman, car les scientifiques savent que le fait de placer l’acide aminé Proline (P) dans des protéines au mauvais endroit entraîne souvent des protéines à devenir non fonctionnelles.
Mais d’autres changements d’acides aminés n’ont pas d’importantes conséquences. Par exemple, changer l’acide aminé alanine (A) pour la thréonine (T) n’est généralement pas nocif pour la fonction protéique. Alors, comment décidons-nous si le changement est nuisible (changer «encode» pour «explode» dans notre phrase) ou un polymorphisme (changer «encode» pour «makes» dans notre phrase qui ne changerait pas le sens)? Premièrement, le gène UBE3A devrait être séquencé chez les parents et/ou chez tout membre de la famille disponible. Pour comprendre comment ce type d’analyse nous dirait si le changement cause le syndrome d’Angelman, examinez ces scénarios ci-dessous. Dans tous ces cas, une mutation ou une «faute d’orthographe» est découverte dans le gène UBE3A d’un enfant et les parents et / ou frères et sœurs sont ensuite testés :
- Aucun parent n’a la même erreur dans son gène UBE3A. Si l’enfant répond aux critères cliniques du syndrome d’Angelman, nous supposerons qu’il ou elle a le SA causé par une mutation aléatoire UBE3A et ce résultat du test serait probablement positif pour le SA.
- Le gène UBE3A de la mère est normal mais le père a la même mutation que l’enfant. Parce que l’UBE3A du père est «silencieux» sur le chromosome de son enfant, nous supposons que cette mutation est un polymorphisme inoffensif. Ce résultat de test serait négatif pour le SA.
- L’UBE3A de la mère a la même mutation que celle de l’enfant, mais ses autres enfants ont également la même mutation mais ne répondent pas aux critères cliniques du syndrome d’Angelman. Nous supposons que cette mutation est un polymorphisme inoffensif. Ce test serait négatif pour le SA.
- Le gène UBE3A de la mère a la même mutation que celui de l’enfant, mais l’UBE3A du père est normal. Les parents de la mère sont testés et la grand-mère maternelle a également cette mutation. Nous supposons qu’il s’agit d’un polymorphisme inoffensif car la mère a hérité de la mutation de sa mère mais n’a pas elle-même la SA. Ce résultat de test serait négatif pour le SA.
- Le gène UBE3A de la mère a la même mutation que celui de l’enfant mais celui du père est normal. Les parents de la mère sont testés et son père a la même mutation. Nous supposons que l’enfant a le SA causé par une mutation héréditaire du gène UBE3A qui provient du grand-père maternel. Tous ses enfants ont 50% de chances de porter cette mutation génique mais elle ne provoquerait pas du SA chez ses propres enfants puisque son UBE3A n’est pas exprimé dans sa progéniture. Cependant, toutes ses filles pourraient avoir des enfants atteints du SA car cette copie mutée sera héritée par leurs enfants de leur mère. On suppose que la mère de l’enfant affecté a 50% de chances d’avoir plus d’enfants atteints du SA. Le test du gène UBE3A est indiqué pour tous les enfants du grand-père maternel pour voir s’ils ont également hérité de cette mutation génétique.
- Au fur et à mesure que de plus en plus d’individus porteurs de mutations UBE3A sont découverts, nous constaterons également que plusieurs individus ont les mêmes changements de séquence dans UBE3A. Si plusieurs personnes non apparentées qui correspondent aux critères cliniques du syndrome d’Angelman ont le même changement de séquence, et que ce changement n’est pas présent chez les personnes qui ne sont pas atteintes du SA, il est probable que ce changement affecte la fonction de l’UBE3A. Ce résultat de test serait positif pour le SA.
Cet article a été rédigé par Rebecca D Burdine PhD et Erin Sheldon et revu par Wen-Hann Tan, BMBS Children’s Hospital Boston, Boston MA. La traduction a été réalisée par FAST France.